








1. 서론
Gardenia jasminoides(GJ)는 일반적으로 Cape jasmine으로 알려져 있으며, Rubiaceae과에 속하는 상록 관목이다(Chen 등, 2020). 전통적으로 동아시아 의학 특히 중국, 일본, 한국에서 사용되었으며 천연 황색 염료로 사용되었을 뿐만 아니라 항염증(Hyun 등, 2008), 항산화(Debnath 등, 2011), 수면의 질 향상(Kuratsune 등, 2010) 및 항우울 활성(Tao 등, 2014)을 포함한 치료적 특성으로 인해 높이 평가되었다. GJ의 여러 화학성분이 분리되고 특성화되었으며 iridoids, iridoid glycosides, triterpenoids, organic acids, flavonoids, 그리고 essential oils과 같은 생리활성 화합물이 풍부하여 광범위한 약리학적 기능에 기여한다(Yang 등, 2009). 약용식물은 수세기 동안 화장품 및 치료제의 활성 성분으로 널리 사용되어 왔으며, 이는 기능성 소재의 잠재적 개발을 위한 알려지지 않은 소스를 제공한다. 또한 화학물질을 사용하지 않고도 건강한 피부를 유지하려는 사람들의 욕구가 증가함에 따라 성장하는 시장 부문이다(Perera 등, 2016). 최근의 식품소재 연구에서는 Gardenia jasminoides seed(GJS) 추출물이 다양한 생리활성을 보유하고 있음을 보고하여 주목받고 있으며, 특히, 피부 건강에 미치는 영향이나 이의 관심도가 급증하고 있다(Park 등, 2019). Tyrosinase와 elastase는 피부 생리학 및 병리학에서 중요한 역할을 하는 효소이다. Tyrosinase(EC 1.14.18.1)는 tyrosine을 멜라닌 생합성의 핵심 효소로 monophenols의 hydroxylation와 O-diphenols의 O-quinones으로의 oxidation을 촉진하고, 이후 중합되어 멜라닌 색소를 형성한다(Prota, 1988). 멜라닌은 피부, 머리카락, 그리고 눈동자 등에서 색을 형성하고 결정 짓는 역할을 한다. 하지만 과도한 멜라닌이 생성될 시, 색소침착(pigmentation)이 생기고, 이로 인한 주근깨(freckles), 반점(blemishes), 혹은 기미(dark spots) 등이 생성될 수 있다(Brozyna 등, 2007). Elastase (EC 3.4.4.7)는 피부의 탄력성을 부여하는 단백질인 엘라스틴(elastin)을 분해하는 효소이다. 하지만 엘라스틴의 분해가 되면 피부의 탄력이 감소하고 주름이 형성되어 피부 노화를 촉진한다. 과도한 elastase의 활동은 만성 염증 및 퇴행성 피부 질환을 포함한 다양한 병리학적 상태와 관련이 있다(Kim 등, 2006). 이러한 질환의 발병과 진행에는 다양한 요인이 관여하지만 그중 산화스트레스는 핵심적인 원인이다(Rinnerthaler 등, 2015). Kelch-like-erythroid cell-derived protein with cap‘n’collar homology-associated protein 1(KEAP1)-nuclear factor erythroid-2-related factor 2(NRF2) 시스템은 세포의 산화 스트레스 반응에 중심적인 조절자로 세포질 내에서 Nrf2와 결합하여 단백질 복합체를 형성함으로써 Nrf2의 분해를 유도하고, 이로 인해 항산화 유전자 반응이 억제된다. 그러나 산화 스트레스나 전자전달계의 이상 등 다양한 자극이 있을 때 KEAP1은 구조적 변화로 인하여 Nrf2가 핵으로 이동하고, 항산화 효소를 포함한 관련 유전자 발현을 촉진한다(Zhang, 2006). KEAP1-Nrf2 pathway의 조절은 피부노화, 그리고 염증성 피부질환과 관련된 연구에서 중요한 기능을 강조하고 피부 항상성에 중요한 역할을 한다. KEAP1의 기능은 피부손상 및 노화를 방지하는데 중요한 역할을 하며, tyrosinase와 elastase의 조절을 통해 피부 보호 및 anti-aging 연구에 관련이 있다(Finkel 등, 2000).
생물정보학의 관점에서 분자도킹(molecular docking)은 컴퓨터를 활용하여 화합물인 리간드(ligand)와 효소(enzyme)의 상호작용을 모델링하는 시뮬레이션의 한 기법으로서, 이 기술로 in vitro, in vivo 연구의 제한점으로 거론되는 비용(money), 시간(time), 인력(manpower)을 감소시킬 수 있다. Molecular docking은 과거 3년 동안 코로나의 여파로 직접적인 스크리닝(식물추출물 활성물질 탐색, COVID-19 저해물질 탐색)의 대체 기법으로 적극 사용이 권장되어 왔으며, 도킹의 정확도 향상, 분자도킹-시험관 내 시험 연관성 규명, AI의 적용 등 지속적인 연구 어프로치가 절실히 필요한 실정이다. 따라서, 본 연구에서는 GJS의 생리활성물질 중 tyrosinase와 elastase에 대한 이중 억제효과(dual inhibitory effect)를 분자 도킹(molecular docking)으로 비교 분석하였다. 이러한 접근 방식은 후속 in vitro 및 in vivo 실험을 위한 유망한 후보 물질을 식별하는데 도움이 될 것이며 궁극적으로 본 연구는 색소 질환 및 피부 노화에 대한 효과적인 식품소재 스크리닝을 통해 화장품 및 식의약 소재 개발을 위한 이들 식품성분의 생체 이용률(bioavailability)의 자료를 확보하는데 그 의의를 둘 수 있다.
2. 재료 및 방법
치자나무 씨앗 추출물의 LC-MS 결과(Saravankumar 등, 2021)를 토대로, 다음과 같은 화합물을 분자도킹에 설정하였다. Iridoids계의 geniposidic acid(GPA), genipin(GNP), geniposide (GPS), genipin gentiobioside(GGB), gardenoside(GDS), shanzhiside methyl ester(SME), 6β-hydroxygeniposide(HGP), coumaroylgenipin gentiobioside(CGG), feruloylgenipin gentiobioside(FGP), monoterpenoides계의 jasminoside F(JAF), jasminoside D(JAD), jasminoside B(JAB) jasminoside J(JAJ), jasminodiol(JOD), gardenate A(GDA), picrocrocinic acid(PCA), jasminoside H (JAH), crocusatin C(CCC), jasminoside A(JAA), 6′-Sinapoyljasminoside C(JSC), methyl dihydrojasmonate(MDJ), 2-hydroxyethylgardenamide A(HEA), flavonoids계의 rutin(RUT), quercetin-3-O-β-D-glucopyranoside(QBG), carotenoids계의 crocetin(CCT), crocin A(CRA), 유기산 및 기타물질로 quinic acid(QNA), trimethoxy-O-glucopyranoside(TOG), 4-(2-Hydroxyethyl)-2-methoxyphenyl β-d-glucopyranoside(HEG), caffeoylquinic acid(CQA), protocatechuic acid(PTA), chlorogenic acid(CGA), dicaffeoylquinic acid(DQA), linolenic acid(LNA), n-pentadecanal(PDA), linoleic acid(LLA), acetylursolic acid (AUA), palmitic acid(PMA), ethyl palmitate (EPA)를 후보물질로 선정하였고 control로 arbutin과 epigallicatechin-3-gallate (EGCG)가 사용되었다.
본 연구에서 분자 도킹 분석은 널리 사용되는 프로그램인 AutoDock Vina(version 1.1.2, The Scripps Research Institute, La Jolla, CA, USA)를 이용하여 수행하였다(Trott 등, 2010). 도킹에 사용할 단백질 구조는 Protein Data Bank(PDB)에서 확보하였으며(Burley 등, 2017), 구조 내 불필요한 잔기 및 공동결정된 리간드는 제거한 후, 에너지 최소화 과정을 통해 구조를 안정화시켰다. 리간드는 PubChem에서 다운로드하였으며, 동일하게 에너지 최소화를 진행한 후 사용하였다. 그리드 박스는 해당 단백질의 active site를 포함하도록 설정하였으며, 주요 결합 잔기에 대한 정보는 관련 문헌을 참고하여 확인하였다. 도킹 분석은 AutoDock Vina와 함께 Molegro Virtual Docker (MVD) (version 6.0, Molegro, Aalborg, Denmark)를 병행하여 수행하였다. 도킹 전 모든 구조에서 물 분자 및 리간드를 제거하였으며, tyrosinase와 elastase의 초기 구조에는 극성 수소 원자를 추가하였다. 그리드 박스의 중심 좌표는 tyrosinase의 경우 x = -10.02, y = -28.82, z = -43.60, elastase의 경우 x = 14.15, y = 48.15, z = -1.16으로 설정하였다. 박스의 크기는 모두 23 × 23 × 23 Å으로 동일하게 적용하였다. 최종적으로 얻어진 도킹 결과는 PyMOL 및 Discovery Studio Visualizer를 이용하여 시각화하고 분석하였다.
물리화학적 및 약동학적 매개변수, 가능한 대사 및 독성 위험(돌연변이원성, 발암성, 심장독성, 피부자극성, 간독성)은 Protox2 web server와, SwissAdme 프로그램을 사용하였다(Daina 등, 2017), AdmetSAR(Cheng 등, 2012) 및 pkCSM (Pires 등, 2015), 약리학적 특성은 PASS Online(Filimonove 등, 2014)를 사용했다. 생물학적 또는 약리학적 활동 스펙트럼 예측을 위한 온라인 예측 결과는 가능한 활성(Pa) 및 가능한 비활성(Pi)의 백분율로 표시하였다. PASS 온라인은 Pa 및 Pi값이 0.000에서 1.000까지 다양하므로, Pa>Pi 및 Pa>0.7만을 고려했다. 합리적인 약물 설계에 자주 사용되는 리핀스키의 5가지 규칙(Pollastri 등, 2010)도 확인하여 리간드들의 생체이용률을 예측하였다.
Tyrosinase와 elastase를 단백질 데이터베이스에서 RSCB (https://www.rcsb.org/)(PDB ID: 7EST), (PDB ID: 2Y9X)의 3D 구조를 얻었다. PubChem 데이터베이스에서 스크리닝된 화합물의 3D 구조를 SDF(구조 데이터 파일) 형식으로 검색하였다.
3. 결과 및 고찰
Tyrosinase의 억제 효과를 알아보기 위해 치자나무 씨앗으로부터 compound(Table 1)를 AutoDock Vina와 Molegro Virtual Docker프로그램을 사용하여 binding energy(Table 2)를 비교하였다. 그 결과, AutoDock Vina의 컨트롤인 Arbutin은 -6.291의 binding energy값을 나타낸 반면, 각 그룹에서 가장 높은 수치(kcal/mol)는 iridoids 그룹에서 coumaroylgenipin gentiobioside(CGG)가 -8.234, monoterpenoides 그룹의 6′-sinapoyljasminoside C(JSC)가 -7.736, flavonoids 그룹의 rutin (RUT)이 -8.049, carotenoids 그룹의 crocin A(CRA)가 -6.811, 기타 dicaffeoylquinic acid(DQA)가 -8.295로 각각 높은 결괏값을 나타내었다. 이후 AutoDock Vina의 결괏값을 상대적으로 비교하기 위하여 Molergro Virtual Docker 프로그램을 사용하여 tyrosinase에 대한 40가지의 화합물의 docking score를 측정하였다. 한편, Molegro Virtual Docker에서는 각 그룹에서 높은 score로는 arbutin -117.761, feruloylgenipin gentiobioside -192.927, monoterpenoides 6′-sinapoyljasminoside C -7.736, rutin -175.57, crocin A -240.501, dicaffeoylquinic acid -179.461로 각각 나타났다. Molegro Virtual Docker의 수치로는 crocin A가 -240.501로 가장 높은 score임을 확인하였다. 반면, dicaffeoylquinic acid는 AutoDock Vina의 순위와는 달리 상대적으로 낮은 값을 나타내었다. 이러한 결과의 차이는 각 프로그램의 스코링 함수, 탐색 알고리즘, 세분된 매개변수, 유연성 모델링의 차이 때문으로 판단된다(Shamsian 등, 2024). 따라서, 두가지의 프로그램의 결괏값을 이용하여 각각의 AutoDock Vina와 Molegro Virtual Docker의 값을 백분율로 환산해 평균값을 구해보았다(Table 3). 그 결과, tyrosinase에 대한 각 화합물의 binding energy의 백분율 값은 RUT, CGG, JSC, FGP, DQA, AUA, HGP의 순으로 높은 값을 나타내었다.
Elastase의 억제 효과를 확인하기 위해 치자나무 씨앗으로부터 얻은 compound를 AutoDock Vina와 Molegro Virtual Docker 프로그램으로 분석하고 이의 binding energy를 각각 Table 1과 Table 2에 나타내었다. 그 결과, AutoDock Vina의 컨트롤인 EGCG의 결괏값은 -7.791의 binding energy값을 나타낸 반면, 각 그룹에서 가장 높은 수치(kcal/mol)는 iridoids 그룹에서 CGG -7.908, monoterpenoides 그룹의 JSC가 -7.577, flavonoids의 그룹 RUT -8.155, carotenoids 그룹의 CRA가 -7.605, 기타 AUA가 -7.582로 각각 높은 결괏값을 나타냈다. 한편, Molegro Virtual docker의 MolDock score는 각 그룹에서 높은 score로는 FGP, -186.736, JSC -163.887, RUT -178.454, CRA -240.501, DQA -167.728로 나타났다.
두 가지의 프로그램의 결과값을 이용하여 각각의 AutoDock Vina와 Molegro Viartual Docker의 값을 백분율로 환산해 평균값을 구해보았다(Table 3). 그 결과 CRA, RUT, FGP, CGG, JSC 순으로 높은값을 나타냈다.
결과적으로 tyrosinase와 elastase의 백분율을 전체적으로 비교한 결과 공통으로 높은 바인딩 에너지 값을 가진 화합물은 RUT, CGG, FGP로 확인되었다. 이후 도킹 결과를 바탕으로 효소의 활성 사이트에 리간드가 바인딩하는 포즈를 확인하였으며, 나아가 Discovery Studio를 통하여 리간드와 단백질과의 상관관계를 확인하였다(Fig. 1). Tyrosinase의 리간드 상호작용 분석 결과, RUT 화합물은 아미노산 잔기인 HIS85 및 ASN81과 전형적인 수소 결합을 형성하고, VAL283 및 VAL248과는 pi-alkyl 결합을 이루며, PHE264와는 pi-pi T형 결합을, MET257과는 pi-sulfur 결합을 형성하는 것으로 확인된다. 반면, CGG 화합물은 MET280 및 HIS85와 수소 결합을 형성하고, VAL283 및 ALA80과는 alkyl 결합을, CYS83과는 carbon-hydrogen 결합을 이루며, GLU322와는 unfavorable acceptor-acceptor interaction 상호작용을 보인다. CRA 화합물은 ARG268 및 ASN81과 conventional hydrogen bond를 형성하며, PHE264, VAL 248, VAL283, PRO284와 alkyl 상호작용을 갖는다. 반면 HIS85, GLU322와는 carbon hydrogen bond를 확인하는 것으로 나타났다. FGP 화합물의 경우는 MET280, ASN260, HIS85 및 ASN81과 전형적인 수소 결합을 형성하고, HIS263과는 pi-pi 스택 결합을, HIS59, HIS61 및 VAL283과는 Alkyl 결합을 형성하며, GLY281과는 carbon-hydrogen 결합을 이루는 것으로 나타났다. 모든 화합물은 공통적으로 HIS85 잔기가 결합하는 것이 관찰되었다(Fig. 2).
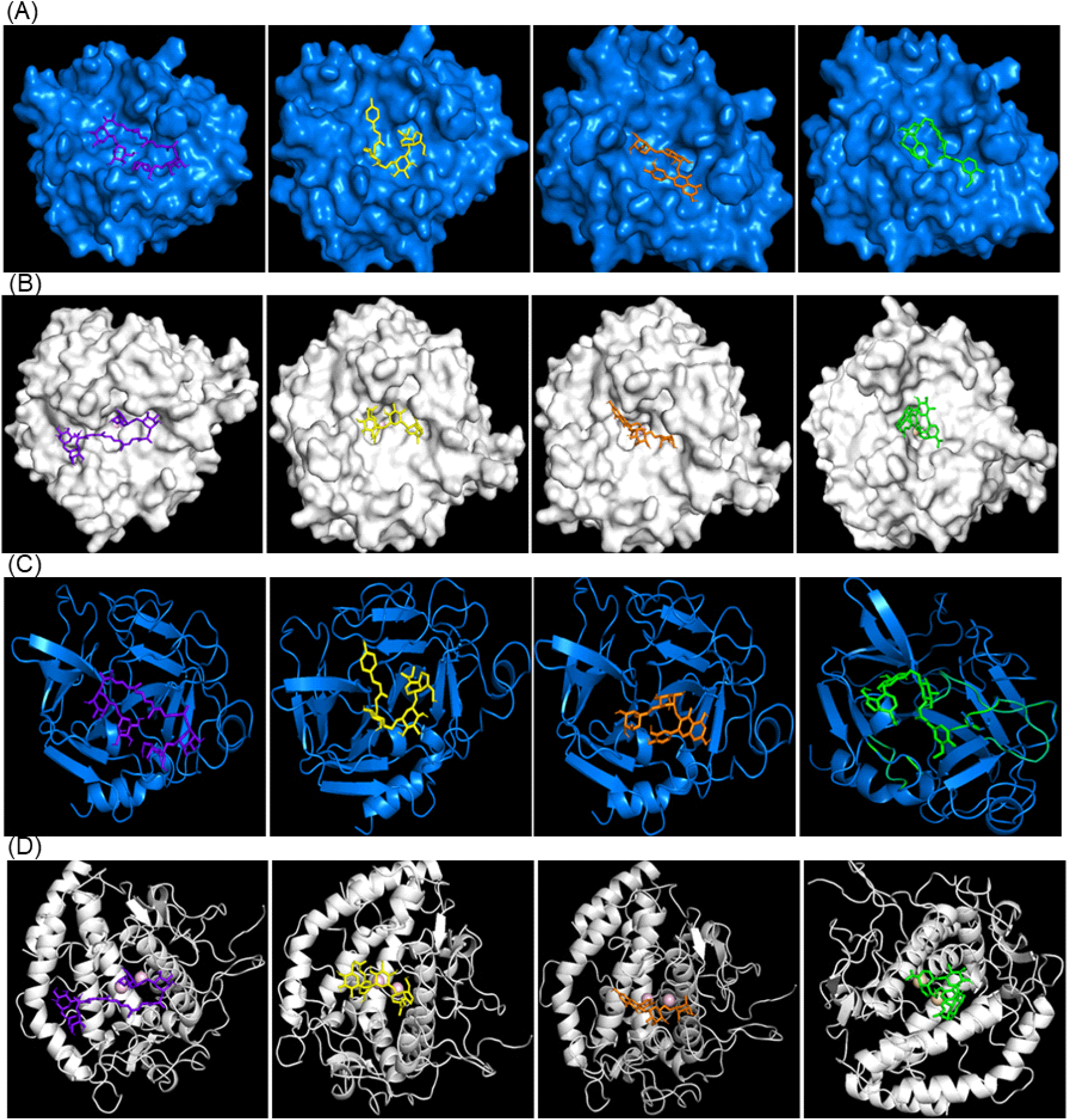
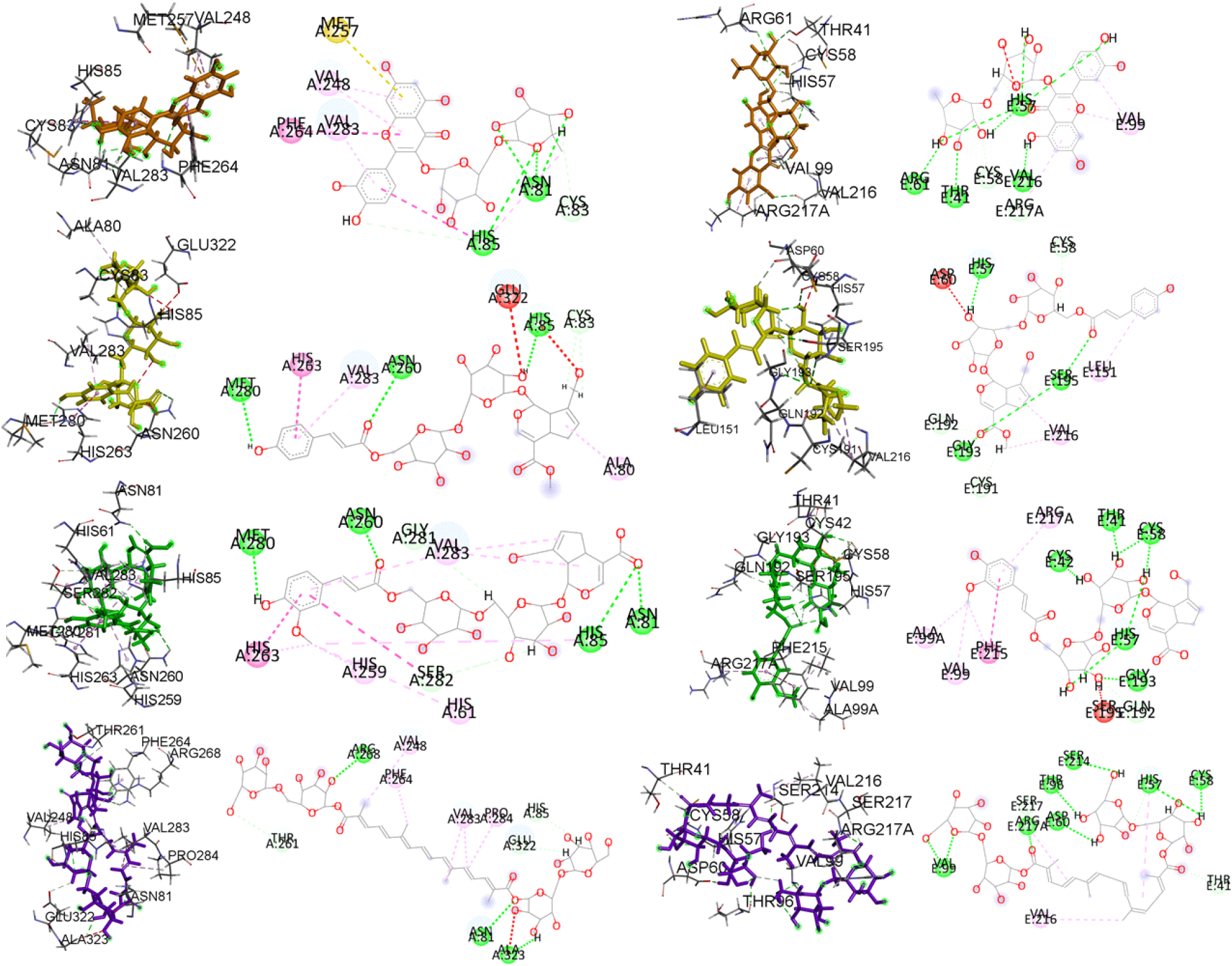
Elastase와 리간드 간의 상호작용 결과, RUT 화합물은 HIS57, VAL216, THR41 및 ARG61과 conventional hydrogen bond를 형성하며, CYS58 및 ARG217과는 carbon-hydrogen bond를 형성하는 것으로 나타났다. 또한, VAL99와는 pi-alkyl 결합을 이루는 것으로 확인되었다. 한편, CGG 화합물은 HIS57, SER195 및 GLY193과 conventional hydrogen bond를 형성하고, CYS58, CYS191 및 GLN192와는 carbon-hydrogen bond를 형성하는 것으로 나타났다. LEU151 및 VAL216과는 alkyl 결합을, ASP60과는 unfavorable acceptor-acceptor interaction을 보인다.
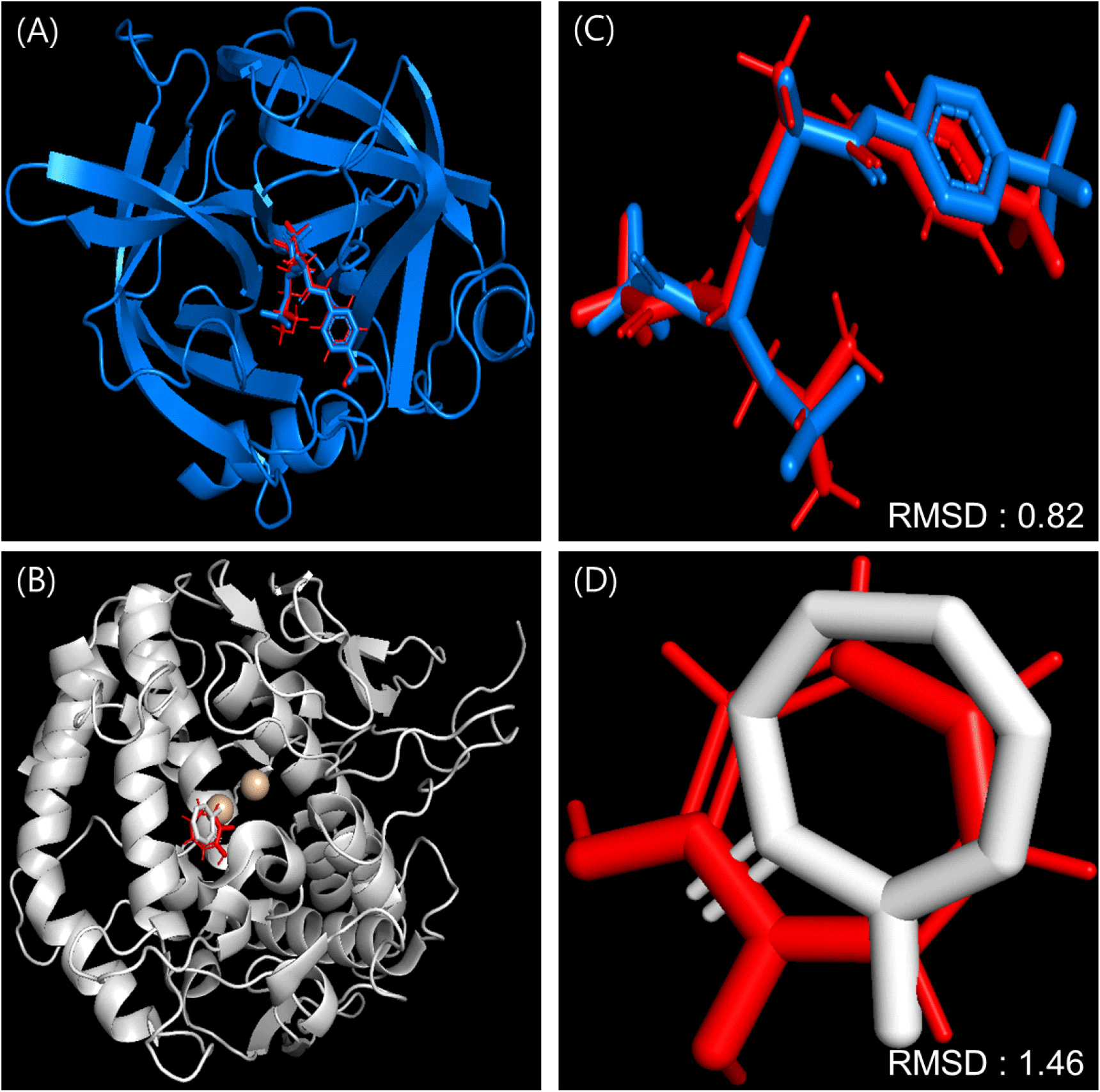
CRA화합물의 경우에는 elastase 상호작용으로는 VAL99, ARG217, ASP60, THR96, SER214, HIS57, CYS58의 잔기와 conventional hydrogen bond를 형성하고 VAL216과 alkyl 결합 및 THR41과 carbon hydrogen bond를 형성하는 것으로 확인되었다. 반면에 FGP 화합물은 ARG217, ALA99 및 VAL99와 alkyl 결합을 형성하며, CYS42, THR41, HIS57, CYS58 및 GLY193과는 conventional hydrogen bond를 형성하고, GLN192와는 carbon-hydrogen bond를, PHE215와는 pi-pi stacked 결합을, SER195와는 unfavorable donor-donor interaction을 이루는 것으로 나타났다. 결과적으로, HIS57과 CYS58은 모든 상호작용에서 공통으로 결합하는 것이 확인되었다. 이러한 도킹 결과는 RMSD를 통하여 신뢰성을 검토하였다. 일반적으로 RMSD(Root Mean Square Deviation) 값은 도킹 프로토콜의 검증, 단백질 안정성 분석, 구조적 변화 예측에 활용된다. RMSD 값은 단백질과 리간드 간의 결합 상호작용과 에너지에 따라 달라지며, 복합체 리간드의 X선 결정 데이터와 시뮬레이션 결과 간 RMSD 값이 2.0 Å 미만일 경우, 변동이 적어 허용 가능한 값으로 간주된다. 본 연구에서는 Molegro Virtual Docker를 이용해 RMSD 값을 0.82Å와 1.46Å로 확인하였으며, 이를 통해 사용한 도킹 프로토콜이 분자 도킹 분석에 적합함을 입증하였다(Fig. 3).
물리화학적, 약리학적 특성 및 생물학적 활성을 포함한 독성 위험을 고려할 때, 선택된 화합물은 특정 기준을 충족해야 한다. 도킹 시뮬레이션 연구를 통해 결합에너지를 확인한 후, 물리화학적, 약동학적, 독성 및 약리학적 특성을 예측하였다. 이는 표적에 대해 가장 좋은 결합 상호작용을 보이는 화합물이 반드시 최상의 약물이라는 보장이 없기 때문이다. 이 과정에서 pkCSM 및 SwissADME 웹 서버를 활용하여 화합물의 ADMET 프로파일을 평가하였다. 이상적인 경구용 약물은 위장관에서 빠르고 완전하게 흡수되어 표적에 특이적으로 분포하고, 활성이 즉시 제거되지 않도록 대사되며, 손상을 일으키지 않고 적절한 방식으로 제거되어야 한다. 약물동역학의 4대 주제인 흡수, 분포, 대사 및 배설은 약물 실패의 주요 원인이며, 이는 사람에서 발생하는 약동학적 예측 오류가 중요한 역할을 한다는 점을 보여준다. 약물 개발에 있어 화학적 구조와 생리학적 매개변수 간의 관계는 필수적이다. 예를 들어, 극성 표면적(PSA)은 약물 투과성을 평가하는 표면적 설명자로, 질소, 산소 및 이들과 결합한 수소 원자가 기여하는 표면적 부분으로 정의된다. 또한, 분자량도 중요한 설명자로, 분자가 클수록 수소 결합을 형성할 수 있는 작용기가 많아지며, 일반적으로 분자량이 500 Da를 초과하는 화합물은 10개 이상의 회전 가능한 결합을 가질 가능성이 높다. 옥탄올-물 분배 계수(logP)는 약물의 소수성을 나타내는 주요 물리화학적 매개변수로, 친유성이 높은 화합물은 대사가 증가하고 흡수가 저하되며, 원치 않는 소수성 거대분자와 결합할 가능성이 커져 독성 잠재력이 높아진다. 이러한 매개변수는 허용 가능한 활성을 평가하는 데 중요한 역할을 한다(Veber 등 2002). Table 3에 나타난 바와 같이, 39가지 MS 데이터를 기반으로 한 화합물을 SwissADME 서버로 분석하여 리핀스키 규칙을 확인한 결과CGG, CRA, FGP, RUT를 포함하여 도킹에서 높은 결합 능을 보인 16가지 화합물이 리핀스키 규칙을 위반하는 것으로 확인되었다(Table 4). 본 연구에서는 리핀스키 규칙에 따른 약리학적 특성을 분석한 결과, CGG번 화합물은 분자량 696.65 g/mol로, 500 g/mol 이하의 기준을 초과하며 49개의 중원자와 6개의 방향족 중원자를 포함하고, 13개의 회전 가능한 결합을 가져 분자의 유연성을 증가시켜 약리학적 활성을 개선할 가능성이 있다. 수소 결합 수용체는 17개, 공여체는 8개로 수용성과 생체막 투과성을 향상시키며, 매우 높은 수용성(very soluble)으로 분류되지만 장내 흡수율은 24.972%로, 생체이용률은 0.11로 상대적으로 낮고, CYP2D6 기질 및 억제제가 아니므로 대사 안정성이 기대되며, Caco2 투과성은 0.264 log Papp in 106 cm/s로 BBB(혈액-뇌 장벽)를 통과하지 못하고, 피부 투과성은 -12.49 cm/s로 피부 흡수에 제약이 있다. 2번 화합물은 분자량 977.0 g/mol로 기준을 초과하고, 18개의 회전 가능한 결합, 24개의 수소 결합 수용체 및 14개의 공여체를 가지며, 물에 잘 용해되나 장내 흡수율은 0%로 실제 흡수에 제약이 있으며, 생체이용률은 제공되지 않았고, Caco2 투과성은 -1.408 log Papp in 106 cm/s로 BBB 통과 가능성이 높지만 피부 투과성은 -2.735(log Kp)로 제한적이다. 3번 화합물은 분자량 712.65 g/mol로 기준을 초과하고, 50개의 중원자, 6개의 방향족 중원자, 13개의 회전 가능한 결합을 가지며, 수소 결합 수용체는 18개, 공여체는 9개로 수용성과 생체막 투과성을 증진시키나 장내 흡수율은 0%로 실제 흡수에 제약이 있고, 생체이용률은 0.11로 낮으며, Caco2 투과성은 -0.785 log Papp in 106 cm/s로 BBB 통과에 실패하고, 피부 투과성은 -12.84 cm/s로 흡수가 어렵다. 마지막으로 4번 화합물은 분자량 610.52 g/mol로 기준에 부합하지만 높은 수치를 보이며, 43개의 중원자, 16개의 방향족 중원자, 6개의 회전 가능한 결합을 가지며, 수소 결합 수용체는 16개, 공여체는 10개로 수용성과 생체막 투과성에 긍정적인 영향을 미치고, ‘용해 가능(soluble)’으로 분류되며, 장내 흡수율은 23.446%로 효과적인 흡수가 가능할 것으로 예상되나 생체이용률은 0.17로 낮고, Caco2 투과성은 -0.949 log Papp in 106 cm/s로 BBB를 통과하지 못하고, 피부 투과성은 -10.26 cm/s로 흡수에 제약이 있다. 분석한 화합물들은 리핀스키 규칙에 따라 각각의 물리화학적 특성과 생물학적 활성을 평가한 결과, 대체로 높은 분자량과 회전 가능한 결합 수, 수소 결합 수용체 및 공여체 수를 가지며, 이는 약리학적 활성을 개선할 가능성이 있는 요소로 작용할 수 있음을 보여준다. 그러나 장내 흡수율이 낮거나 0%인 화합물들이 존재하여 실제 흡수에 대한 제약이 있으며, 일부 화합물은 BBB(혈액-뇌 장벽) 통과 가능성이 있는 반면, 피부 투과성이 매우 제한적이라는 점에서 피부를 통한 약물 전달이 어려운 것으로 나타났다. 따라서, 이러한 물리화학적 특성들은 화합물의 생리적 환경에서의 약리학적 활성을 발휘할 가능성을 평가하는 데 중요한 기초를 제공하며, 향후 추가적인 최적화 및 구조적 수정이 필요할 것으로 판단된다.
1) CGG, Coumaroylgenipin gentiobioside; CRA, crocin A; FGP, feruloylgenipin gentiobioside; RUT, rutin.
1) Web server: pkCSM (http://biosig.unimelb.edu.au/pkcsm), and swissAdme (http://www.swissadme.ch/) and admetSAR (https://lmmd.ecust.edu.cn/admetsar1/home/).
본 연구에서는 다양한 생리활성을 예측하기 위해 PASS (Prediction of Activity Spectra for Substances)-online 웹 서버를 사용하였다(Table 5). PASS-online은 화합물의 구조 정보를 기반으로 생리활성 가능성을 예측하는 도구로, 다양한 화학적 구조에 대해 약리학적 활성을 높은 정확도로 예측할 수 있다. 이를 통해 분석한 CGG, CRA, FGP, RUT 화합물의 활성 프로파일을 바탕으로 tyrosinase와 elastase 억제와 관련된 가능성을 평가하였다. CGG 화합물은 free radical scavenger와 anti-inflammatory 특성을 통해 산화 스트레스와 염증 반응을 억제하여 세포막의 안정성을 증가시킬 것으로 예상된다. 또한, membrane permeability inhibitor로서 피부 보호 및 anticarcinogenic 활성을 발휘하여 세포 손상을 예방하는 데 기여할 수 있으며, apoptosis antagonist와 immunosuppressant 활성을 통해 세포 자멸을 억제하고 면역 반응을 조절함으로써 피부 건강에 긍정적인 영향을 미칠 가능성이 있다. 이러한 특성들은 tyrosinase와 elastase 억제 작용을 보조할 수 있는 중요한 요소로 작용할 수 있다. CRA 화합물은 항산화 기능을 통해 활성 산소종(ROS)을 중화시키고, elastase와 tyrosinase의 과도한 활성을 억제하여 결합 조직 보호, 미백 및 항노화 효과를 나타낼 수 있을 것으로 보인다. 또한, 염증 반응에서 elastase에 의한 세포외 기질 분해와 조직 투과성 증가를 방지하여 조직을 보호하고 염증을 줄이며, CYP2E1 유도제로 산화 반응을 조절하여 활성 산소를 감소시키고, 자유 라디칼 소거제로서 산화 손상을 억제하는 방식으로 elastase와 tyrosinase의 활성을 조절할 가능성이 있다. 더불어, 세포 자멸 유도 특성은 산화 스트레스로 손상된 세포를 제거하여 효소의 부정적 영향을 줄일 수 있음을 시사한다. FGP 화합물은 anti-inflammatory 작용으로 염증 반응을 완화하고, anticarcinogenic 특성으로 세포 손상을 방지하며, antiprotozoal 활성으로 미생물로부터 피부를 보호할 수 있을 것으로 추정된다. 또한, caspase 3 stimulant 특성으로 세포 자멸 과정을 촉진하여 피부 재생을 도울 수 있으며, 이는 tyrosinase 억제 효과와 관련이 있을 가능성이 있다. 마지막으로, RUT 화합물은 free radical scavenger 및 lipid peroxidase inhibitor로서 산화 스트레스를 줄이고, membrane permeability inhibitor 및 membrane integrity agonist로서 피부 장벽을 강화할 수 있다. 또한, anticarcinogenic 및 vasoprotector 효과로 세포 손상을 방지하고 피부 혈관을 보호하여 피부 건강을 유지하는 데 기여할 수 있으며, CYP1A1 inducer로서 피부 대사를 조절하여 tyrosinase와 elastase 억제에 간접적으로 기여할 수 있다. 또한, anti-inflammatory 및 antioxidant 특성으로 염증 및 산화 반응을 줄여 피부 노화를 방지할 수 있을 것으로 예상된다 (Table 6).
1) CGG, Coumaroylgenipin gentiobioside; CRA, crocin A; FGP, feruloylgenipin gentiobioside; RUT, rutin.
1) Web server: (https://www.way2drug.com/passonline/).
1) CGG, Coumaroylgenipin gentiobioside; CRA, crocin A; FGP, feruloylgenipin gentiobioside; RUT, rutin.
1) Web server: (https://tox.charite.de/protox3/index.php?site=home).
화합물 CGG, CRA, FGP, RUT의 독성 프로파일을 Protox2 서버를 활용하여 평가한 결과, CGG와 FGP는 심독성과 면역독성에서 활성으로 나타났으나, 간독성, 신경독성, 돌연변이 유발성 및 세포독성에서는 비활성으로 확인되었다. RUT는 모든 독성 항목에서 비활성으로 상대적으로 안전성을 보였으나, 면역독성에서는 활성으로 나타나 주의가 필요하다. CRA는 간독성, 신경독성, 면역독성에서 비활성으로 나타났으나, 심독성에서는 활성으로 확인되었고, 다른 독성 항목에서는 비교적 안전한 특성을 보였다. 이를 바탕으로, CGG와 FGP는 심독성과 면역독성 문제를 관리할 경우 유망한 약리 효능을 기대할 수 있으며, RUT는 상대적으로 높은 안전성을 보였다. 이러한 독성 평가 결과는 화합물의 독성에 대한 이해를 돕고, 향후 약물 개발에 중요한 참고자료가 될 수 있다.
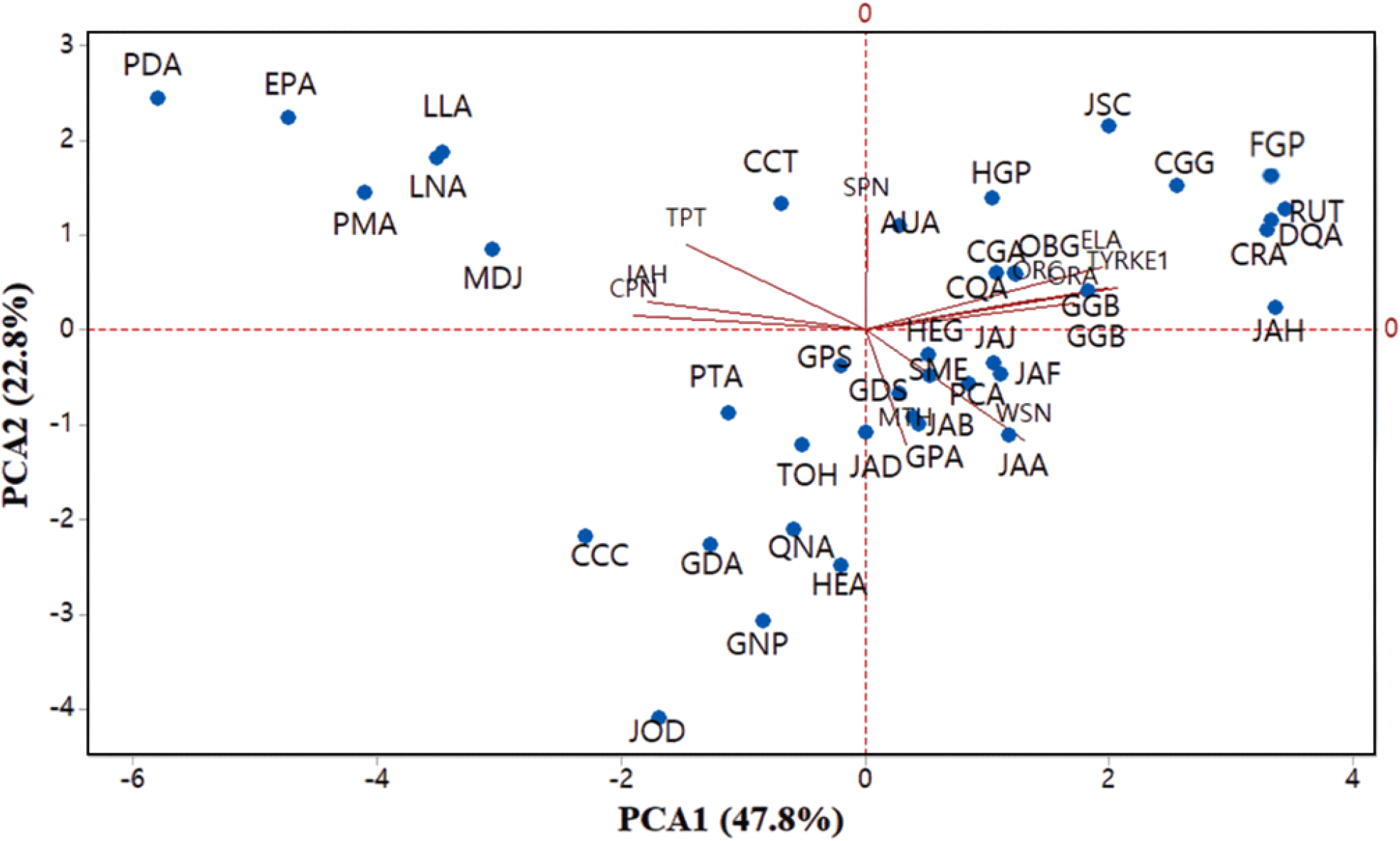
Tyrosinase와 elastase의 이중 활성 억제 기능에 더해, 이전 연구에서 사용한 실험 방법(Diniyah 등, 2023)으로 항산화 능력을 평가하기 위해 Keap1과의 도킹 결합 에너지를 확인하였으며, 결과는 Table 2에 제시되었다. 이후 도킹 결과와 pkCSM 서버를 통해 예측한 약물적 흡수율과 독성 결과를 바탕으로 PCA와 Heatmap 분석을 진행하였다. 예측 항목으로 사용된 팩터는 Water Solubility(WSN; log mol/L), Caco2 Permeability (CPN; log Papp in 10-6 cm/s), Intestinal Absorption(human; IAH; % Absorbed), Skin Permeability(SPN), Max. Tolerated Dose(human; MTH; log mg/kg/day), Oral Rat Acute Toxicity (ORA; LD50; mol/kg), Oral Rat Chronic Toxicity(ORC; LOAEL; log mg/kg_bw/day), T. pyriformis Toxicity(TPT; log μg/L) 등이 포함되었다. 최종적으로 tyrosinase, elastase, Keap1, 그리고 흡수율 및 독성 예측 결과를 바탕으로 PCA와 heatmap 분석을 수행하였다. PCA는 다변량 데이터를 두세 개의 주요 성분으로 차원 축소하여 정보를 최소화하는 도구로 활용되었으며, 전체 변동성은 70.6%였고, PC1은 47.8%, PC2는 22.8%를 차지하였다(Abdi와 Williams, 2010). Tyrosinase, elastase, Keap1의 벡터와 CGG, CRA, FGP, RUT 샘플이 군집하여 해당 변수들이 서로 높은 상관 관계를 가진다는 것을 확인할 수 있었다. Heatmap의 범위는 상관관계가 가장 높을수록 붉은색(3.9)을 나타내고, 가장 낮을수록 파란색(-3.7)을 나타내도록 설정되었다(Pang 등, 2022). 해당 결과에서 RUT은 tyrosinase, elastase, Keap1과 가장 높은 상관관계를 보이며 붉은색을 띠는 반면, CRA는 상대적으로 낮은 elastase와 Keap1과의 상관관계를 보였다(Fig. 4).
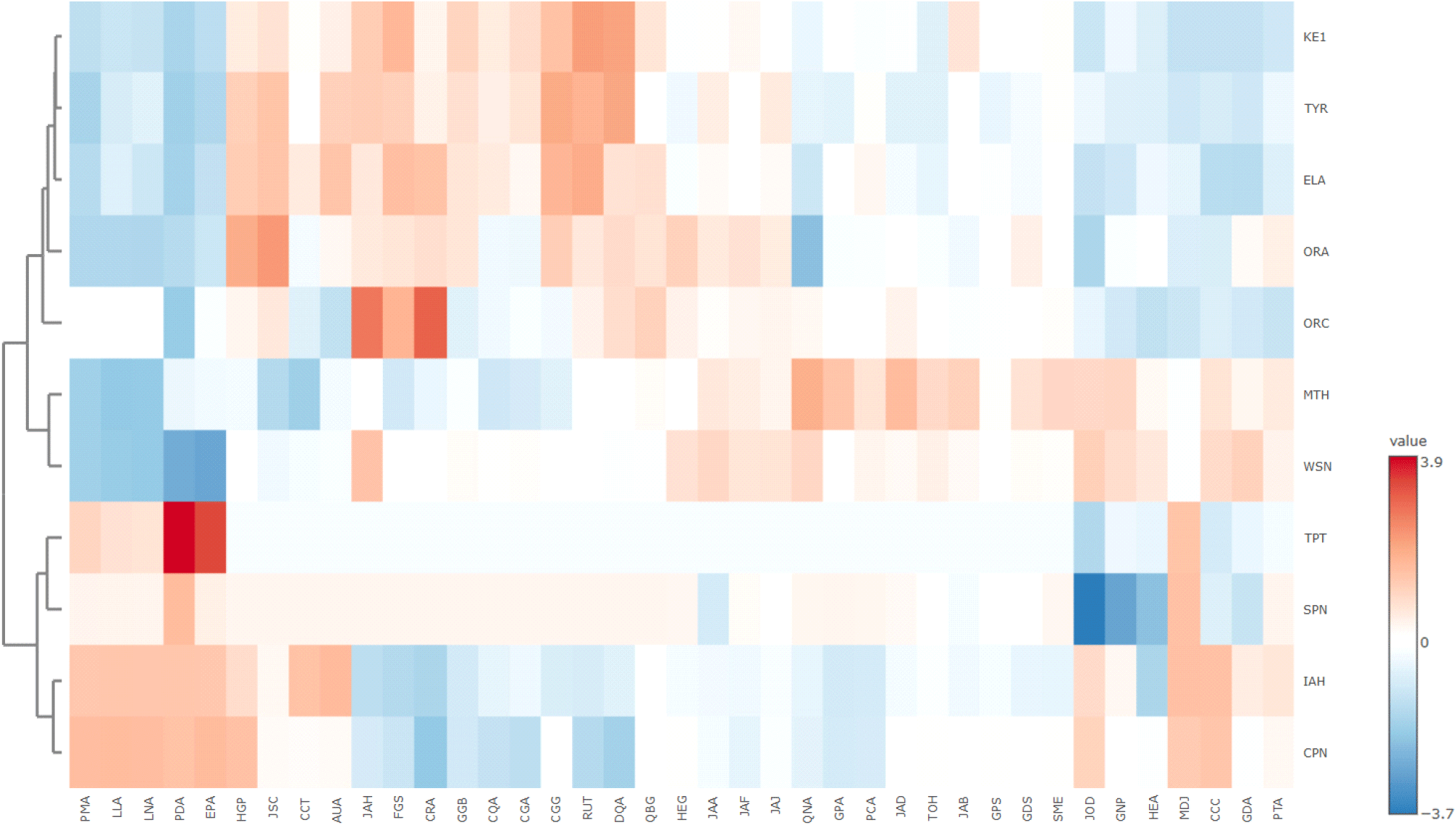
주성분 분석 결과(Fig. 5), CGG, RUT, CRA, FGS는 PCA1 및 PCA2 축에서 모두 양의 값을 나타내며 1사분면에 위치하였다. 이들은 Tyrosinase, KEAP1, Elastase 결합능을 변수로 포함한 PCA 벡터 방향과 유사한 위치에 분포함으로써, 해당 효소들에 대한 높은 결합능 또는 억제 활성을 보일 가능성이 시사된다. 또한, ORA(oral rat acute toxicity, 경구 급성 독성) 및 ORC(oral rat chronic toxicity, 경구 만성 독성) 변수 벡터와도 일치하는 방향성을 나타내, 이들 샘플이 각각 급성 및 만성 독성 측면에서도 상대적으로 높은 값을 가질 가능성이 있음을 보여준다. 특히 CGG는 벡터의 연장선상에서 가장 멀리 떨어져 있어 관련 효능 및 독성 모두에서 강한 특성을 가질 수 있으며, RUT와 CRA 역시 유사한 효능-독성 프로파일을 공유하는 것으로 보인다. 이러한 결과는 해당 물질들이 피부 효소 억제 측면에서는 유망하지만, 동시에 급성 및 만성 독성에 대한 평가가 추가로 필요함을 시사한다.
본 연구에서는 tyrosinase와 elastase의 효소 억제 활성을 동시에 가진 화합물을 발굴하기 위해 인실리코 도킹 기법을 활용하였다. Tyrosinase와 elastase는 각각 멜라닌 생합성과 콜라겐 분해에 중요한 역할을 하는 주요 피부 노화 관련 효소로, 두 효소의 활성을 억제함으로써 미백 및 항노화 효과를 기대할 수 있으며, 이는 다양한 피부 문제 해결을 위한 잠재적 약물 후보 발굴의 기초가 된다.
인실리코 도킹은 여러 가지 장점을 제공한다. 첫째, 시간과 비용 절감의 이점이 있으며, 실험적 방법으로 모든 화합물을 시험하는 것에 비해 가상 환경에서 신속하게 평가할 수 있다. 둘째, 대규모 화합물 라이브러리에서 효소 억제 활성을 예측해 초기 스크리닝 단계에서 유용하게 활용할 수 있다. 셋째, 분자적 상호작용 정보를 제공하여 특정 화합물이 tyrosinase와 elastase에 결합할 때의 결합 방식과 상호작용하는 잔기들을 시각화 함으로써 작용 기전 연구에 기여할 수 있다. 그러나 인실리코 도킹 기법은 몇 가지 한계도 지닌다. 예측 정확도가 완벽하지 않아 실험적 검증이 필요하며, 단백질의 유연성을 충분히 반영하기 어려워 실제 생체 내 효과를 완전히 예측하지 못할 수 있다. 따라서, 본 연구에서도 tyrosinase와 elastase의 억제에 대한 바인딩에너지만을 고려했으며, 대사 및 독성 여부는 추가적인 실험으로 확인이 필요하나 식품성분으로 발견된 성분이 어느 정도 활성을 보유하는지에 대한 신속한 확인을 위해서는 매우 유용하게 적용 가능하다고 판단된다. 구강 투여 약물의 적합성을 평가하기 위해 리핀스키의 규칙(Lipinski’s Rule of Five)을 적용한 결과, 화합물 RUT, CGG, CRA, FGP 모두 분자량, 수소 결합 공여자 수, 수소 결합 수용자 수 항목에서 규칙을 위반하였다. 예를 들어, 화합물 RUT은 분자량이 610.52 g/mol, 수소 결합 공여자 수 8, 수용자 수 16으로 세 가지 항목을 위반하였고, 화합물 CGG, CRA, FGP도 각각 세 가지 항목을 초과하여 구강 투여 적합성에 한계가 있을 가능성이 확인되었다.
구강 투여 적합성을 높이기 위한 구조적 변형 전략으로는 분자량 감소와 수소 결합 공여자 및 수용자 수의 감소가 있다. 활성에 크게 기여하지 않는 비활성 부분이나 큰 치환기를 절단하여 전체 분자량을 줄이고, -OH, -NH, -COOH 등의 작용기를 -CH3, -F 같은 소수성 그룹으로 대체하거나 에스테르화, 메틸화 등을 통해 수소 결합 수를 줄일 수 있다. 이로써 화합물의 지질 친화성을 높여 위장관 흡수를 촉진할 수 있다. 또한, prodrug 설계 방식(Rauto 등, 2008)으로 비활성 전구체 형태로 개발해 대사 후 활성화되도록 하는 방법도 구강 투여 적합성을 향상하는 데 활용될 수 있다.
본 연구에서는 Protox2 서버를 통해 화합물 CGG, CRA, FGP, RUT의 독성 프로파일을 평가하였다. 화합물 CGG과 FGP는 심독성과 면역독성이 활성으로 나타나 특정 장기와 시스템에 미치는 위험성을 시사하였고, 화합물 RUT은 면역독성을 나타내는 것으로 평가되었다. 독성 문제 해결을 위해 구조적 변형, 항산화제 병용, 서방형 약물 설계 등의 전략이 필요하다(Das 등, 2022). 특히, 화합물 RUT은 상대적으로 안전성이 높아 추가 연구가 요구되며, 화합물 CGG과 FGP는 심독성과 면역독성을 관리할 경우, 유망한 후보 물질로 평가된다. Protox2 서버를 통해 얻은 독성 예측 결과는 향후 안전하고 효과적인 약물 개발의 기초 자료로 활용될 수 있다. 결론적으로, 본 연구는 tyrosinase 및 elastase 이중억제활성을 가지는 식품 유래 성분 발굴을 위해 인실리코 도킹기법이 효과적으로 활용될 수 있음을 시사하엿으며, 향후 연구에서는 in vitro assay를 통한 Michaelis-Menten kinetics 분석, 구조-활성 상관관계(SAR) 분석, 및 signaling pathway 규명 뿐만 아니라, Cryo-EM과 Dynamic Bayesian Network Modelling을 활용한 고해상도의 구조분석을 통해 결합억제에 관여하는 유연한 부위와 신호경로를 실시간으로 규명할 필요가 있다. 이러한 접근은 피부노화 또는 피부트러블과 관련된 효소들을 동시에 억제할 수 있는 효과적인 식품성분 개발에 중요한 기반을 제공할 수 있을 것으로 기대된다.
4. 요약
본 연구에서는 치자나무 씨앗의 질량분석 데이터 기반의 화합물을 활용하여 tyrosinase와 elastase에 대한 이중억제 활성을 분석하고자 AutoDock Vina와 Molegro Virtual Docker를 이용한 분자 도킹을 수행하였다. 분자 도킹은 단백질-리간드 간 상호작용을 예측하는 컴퓨터 기반 분석기법으로, 특정 리간드가 표적단백질의 활성 부위에 결합하는 친화도를 정량적으로 평가할 수 있다. 또한, 화합물의 약리학적 특성 평가를 위해 pkCSM, admetSAR, SwissADME, PASSonline 서버를, 독성 예측을 위해 Protox2 서버를 활용하였다. 예측된 데이터를 바탕으로 PCA 및 Heatmap 분석을 수행한 결과,, CGG, CRA, FGP, RUT 화합물이 이중 활성에 공통적으로 영향을 미치는 것으로 확인되었다. 특히,도킹 분석 결과 바인딩 에너지가 가장 높았던 CGG(TYR 93.57792, ELA 85.95508), CRA(TYR 91.05485, ELA 96.62784), FGP(TYR 91.66767 ELA 86.13069), RUT (tyrosinase 90.95614, elastase 87.10047)는 tyrosinase와 elastase의 이중 억제를 유도할 수 있는 유망한 후보물질임을 시사하였다.