Introduction
Cherry elaeagnus (Elaeagnus multiflora Thunb., Em) belongs to the family Elaeagnaceae. It is cultivated as an ornamental and fruit tree, and it is naturally distributed in the wild (Saeng, 1989). The berries of Em mature in 6-7 months to a characteristic red color, with a sweet and slightly sour taste (Hong et al., 2006; Park, 2004). The leaves, stems, and roots of Em have been traditionally used for medicinal purposes. Consuming Em berries is purported to strengthen the five visceral organs. In addition, the berries are used to treat diarrhea, bleeding, dyspepsia, osteomyelitis, edema, and irregular periods (Hong et al., 2007; Yoon et al., 2007). In China, Em is also used to treat cough, itching, wounds, and even cancer (Kim, 1984). The medicinal value of the fruit of Em has been studied, and it is used in small amounts as herbal medicine. However, similar data on its leaf are limited.
The chemical constituents of Em include carbohydrates (24.1%), crude proteins (1.4%), crude fats (0.4%), and crude ash (2.5%) (Yoon et al., 2007). Meanwhile, the anti-inflammatory activity of Em has been described (Lee et al., 2007), but an immune-enhanceing effect has not been reported.
T helper (Th) cells comprise Th1 and Th2 cells. Th1 cells secrete interleukin (IL)-2 and Th2 cells secrete IL-4 and IL-10. The former are involved in delayed hypersensitivity and the latter in antibody production. Th1 and Th2 cells function exclusively in association with each other, and the cytokines they secrete are important in the immune response. IL-2, also termed T-cell growth factor (Mire-sluis and Thorpe, 1998), is secreted by T cells, natural killer (NK) cells, and lymphocyte-activated killer cells. IL-4, which is also termed B-cell growth factor and B-cell stimulatory factor-1, stimulates DNA synthesis in B lymphocytes. Further, IL-4 is produced mainly by T lymphocytes, mast cells, and basophils. The various activities of IL-4 include surface molecule expression, B-cell proliferation and differentiation, and T-cell activation. IL-10 produced by Th2 cells interferes with cytokine production in Th1 cells. Therefore, initially, this molecule was considered a cytokine synthesis inhibitory factor. However, various activities of IL-10, including inhibitory and stimulatory (Mire-sluis and Thorpe, 1998), were subsequently revealed, as was its production by other cells, including macrophages, activated T cells, and mast cells.
In this study, we aimed to evaluate the therapeutic use of Em leaves by evaluating the antioxidant activity and immune-enhancing effect of a hot aqueous extract of these leaves.
Materials and methods
Folin-Ciocalteu phenol reagent, 2,2-diphenyl-1-picrylhydrazyl radical (DPPH), 2,2’-azino-bis-3-ethylbenzothiazoline-6-sulfonic acid (ABTS), gallic acid, ascorbic acid, and concanavalin A (ConA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Roswell Park Memorial Institute (RPMI) 1640, Hank’s balanced salt solution (HBSS), and phosphate-buffered saline (PBS) were purchased from Gibco (Carlsbad, CA, USA). Mouse IL-2, IL-4, and IL-10 ELISA kits were purchased from R&D Systems (Minneapolis, MN, USA). Cytotox 96 non-radioactive cytotoxicity assay kit (g1780), fetal bovine serum (FBS), and Premix WST-1 Cell Proliferation Assay kit (MK400) were purchased from Promega (Madison, WI, USA), Merck (New York, NY, USA), and TaKaRa Bio, Inc. (Shiga, Japan), respectively.
Dried Em leaves (100 g) were placed in a bottle, followed by the addition of 2 L of distilled water. The sample was extracted at 100°C for 3 h using a reflux extractor. The extract was passed through Whatman No.41 filter paper and the filtrate was evaporated using a rotary evaporator. The evaporator bottom was freeze-dried at −50°C for 48 h using a freeze dryer. Fifteen grams (15%) of the extract powder obtained and was used as the experimental sample.
DPPH radical scavenging activity was measured as previously described (McCune et al., 2002). Briefly, 10 μL of sample solution and 90 μL of methanol were added to wells of a 96-well plate, followed by the addition of 100 μL of 0.3 mM DPPH in ethanol. After incubating the solution in the dark for 30 min at 25°C, the absorbance of the solution was measured at 517 nm using a microplate reader. The absorbance of blank solution containing 10 μL of distilled water and 90 μL of methanol was also measured. The radical scavenging activity was compared with that of vitamin C (ascorbic acid; Sigma-Aldrich) as the positive control. The activity (%) was calculated as follows:
The ABTS radical scavenging activity was measured as previously described (Jeong et al., 2009). PBS (pH 7.4) was used to prepare a 1:1 dilution of 2.45 mM ABTS potassium persulfate (7 mM) dissolved in distilled water. The absorbance of the stock solution was 0.70±0.02. Radial stock solution was placed in the dark for 12-16 h. Aliquots (190 μL) of the stock solution were dispensed and 10 μL of the sample at different concentrations was added to each. Following incubation for 7 min the absorbance was measured. As the positive control, vitamin C was used. The antioxidant activity was determined as the percentage of absorbance of the samples without and with extract. The activity (%) was calculated as follows:
The reducing power was measured using a modification of a previously described method (Oyaizu, 1986). Briefly, 200 μL of each sample received 200 μL each of 200 mM PBS and 1% potassium ferricyanide. The solution was stirred at 50°C for 20 min. Thereafter, 200 μL of 10% trichloroacetic acid was added to the solution and the solution was centrifuged at 13,500 ×g for 10 min. The supernatant was collected. Then, 400 μL of 0.1% ferric chloride was added to 400 μL of the supernatant and the absorbance of the solution was measured at 700 nm. The reducing power was converted to percent value of the absorbance ratio of the treatment and control groups.
The total polyphenol content was analyzed using the Folin-Ciocalteu method, based on the principle that the phenolic materials reacts with phosphomolybdic acid and develops a blue color (Singleton at el., 1999). A mixture was prepared of 1 mL of extract (1 mg/mL), 3 mL of distilled water, and 1 mL of Folin-Ciocalteu reagent. The mixture was reacted at 25°C for 5 min, followed by the addition of 1 mL of 7% NaCO3. The mixture was then reacted in the dark for 2 h and the absorbance of the solution was measured. The total polyphenol content in the sample is expressed as gallic acid equivalent per gram of sample using a standard curve of gallic acid (6.25-100 μg/mL).
Male Balb/c mice purchased from Samtaco (Osan, Korea) were used as experimental animals to evaluate the immune-enhancing effect of the Em leaf extract. The mice were maintained under constant conditions (temperature 22±2°C, humidity 50±5%, and 12-h light/dark cycle) for 1 week.
Spleen cells were aseptically removed from the mice following sacrifice by cervical dislocation. The cells were washed with HBSS and rinsed with gentle rubbing against the back of the syringe piston. The cell suspension was washed twice with culture medium and centrifuged at 3,000 rpm for 10 min. The spleen cells were dispersed in RPMI 1640 and stained with trypan blue. The cells were counted using a hemocytometer. The cell concentration was adjusted to 2×105 cells/mL, and 200 μL of the suspension was dispensed into wells of a 96-well plate. This animal experiment was approved by the Animal Research Ethics Committee of the Natural Resources Research Center (JINR1604).
Six-week-old Balb/c male mice were sacrificed by cervical dislocation. The spleen was aseptically removed from each mouse and crushed through a 100-mesh to obtain single cells. The mononuclear cells were collected and centrifuged three times for 5 min each time at 12,000 rpm to obtain splenocytes. The spleen cells were diluted with RPMI 1640 medium containing 10% heat-inactivated FBS to a concentration of 2×105 cells/mL. One hundred microliters of the suspension was dispensed into wells of a 96-well plate. ConA was added to each well and the plate was incubated at 37°C in a 5% CO2 incubator for 30 min, and then at 37°C for 48 h. WST-1 (water soluble tetrazolium salts) solution was added at a concentration of 5 mg/mL and the plate was incubated in the dark for 2 h. The absorbance of each well was measured at 440 nm.
Splenocytes were treated with ConA (0.1 μg/mL) and 50, 100, and 200 μg/mL Em leaf extract, and then dispensed into wells of 96-well plates at a density of 2×105 cells/mL. After 24 and 48 h, the culture supernatant was collected. After incubation at 4°C for 30 min at 1°C or lower, the cells were flushed with wash solution for 2 h and 200 μL of reagent diluent was dispensed into each well. After 1 h, each sample was washed twice and 100 μL of the culture supernatant was incubated for 2 h at room temperature. The supernatant was washed twice and dispensed (100 μL) into a working detector and incubated at room temperature for 2 h. After two washes, 100 μL of horseradish peroxidase-conjugated streptavidin was added and incubated for 20 sec at room temperature. After washing twice, 100 μL of the supernatant was incubated for a maximum of 40 min. Finally, 50 μL of the solution was used to measure the absorbance at 450 nm.
YAC-1 cells were purchased form the Korea cell line bank. Yac-1 cells and mouse spleen cells were cultured in RPMI 1640 medium containing 10% FBS and 1% penicillin at 37°C. After incubation in a 5% CO2 incubator, 50 μL of BALB/c mouse splenocytes (1×106 and 5×105 cells/mL) and 50 μL of YAC-1 cells (2×105 cells/mL) were seeded in wells of a 96-well plate. Subsequently, 50, 100, or 200 μg/mL of Em leaf extract was added and the plate was incubated at 37°C in a 5% CO2 incubator for 4 h. After adding 10 μL of 10 × lysis buffer 45 min before the end of the reaction, the cells were re-cultured and centrifuged at 2,500 rpm for 4 min. Subsequently, 50 μL of the supernatant was transferred into wells of a 96-well plate, 50 μL of substrate mix was added, and the mixture was incubated in the dark for 30 min at room temperature. After stopping the reaction by adding 50 μL of stop solution to each well, the absorbance of the sample was measured at 490 nm using a microplate reader.
We compared the resulting metabolic activities of the treatment groups and controls using one-way analysis of variance and Dunnett’s multiple-comparison posttest. Differences between groups were considered significant at a p-value <0.05. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Results and discussion
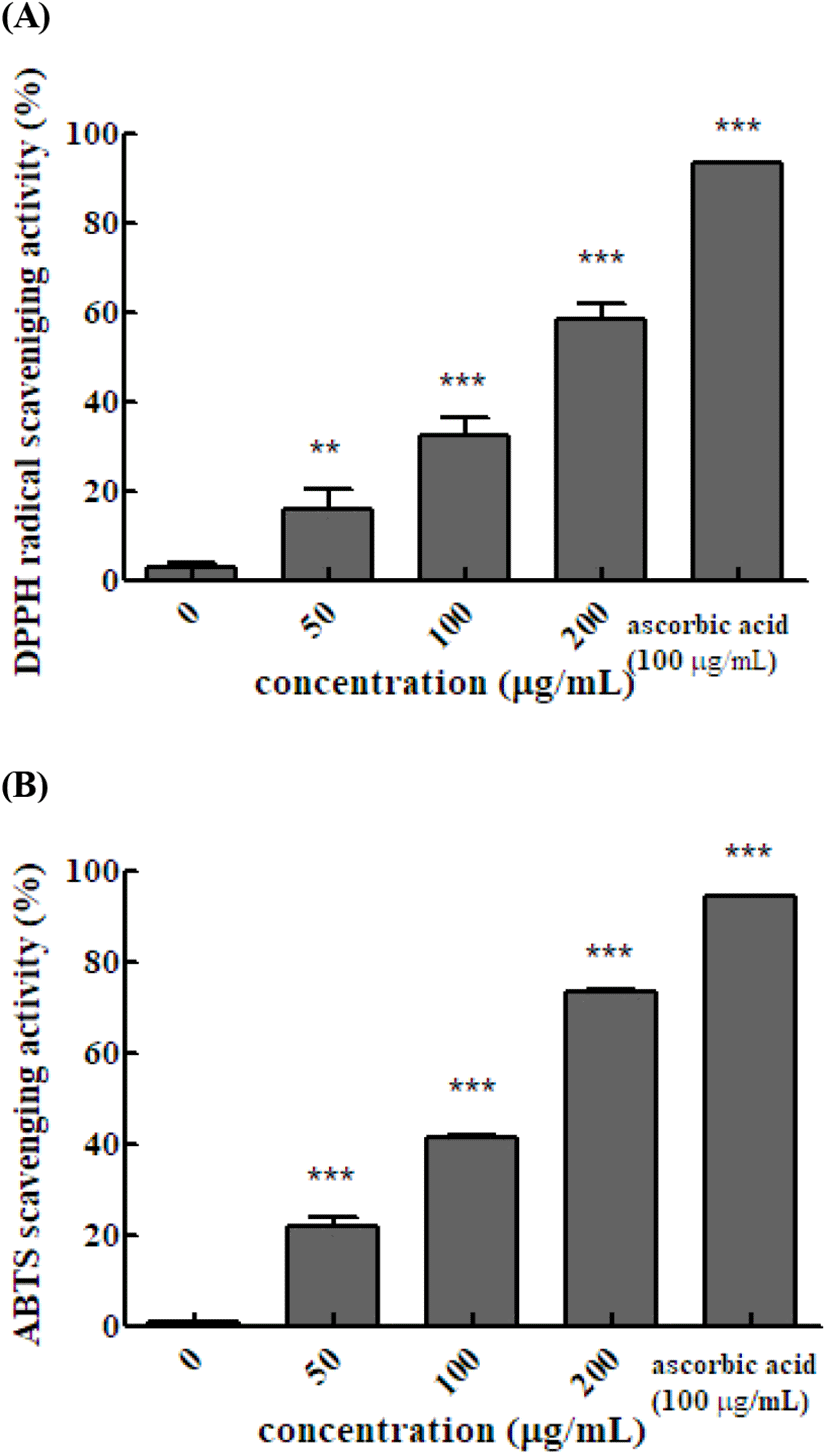
The ABTS and DPPH assays are widely used to measure antioxidant activities using relatively stable free radicals. ABTS produces cation radicals and DPPH produces anion radicals. Therefore, the two methods differ in the degree of binding between the substrate and reactant. and the results differ between the methods. The DPPH radical scavenging activity of the Em leaf extract was 16.6%, 32.5%, and 58.5% at 50, 100, and 200 μg/mL, respectively. The DPPH radical scavenging activity of 100 μg/mL vitamin C was 93.4%. The DPPH radical scavenging activity increased significantly as the concentration of the hot water extract of Em leaves increased. In the ABTS method, the antioxidant activity is measured by exploiting the fact that ABTS produced by reaction with potassium persulfate is removed by the antioxidants in the sample, resulting in discoloration. The ABTS radical scavenging activity with 50, 100, and 200 μg/mL of extract was 22.0%, 41.4%, and 73.2%, respectively, and that of 100 μg/mL vitamin C was 94.6%. The DPPH radical scavenging activity of hexane, dichloromethane, and acetyl acetate fruit extracts of Em has been reported to be 62.92±2.45, 65.25±4.74 and 98.83± 0.02%, respectively (Kim, 2007). The results of the present study confirmed that, similar to the fruits, the leaves of Em also exhibit DPPH radical scavenging activity (Fig. 1).
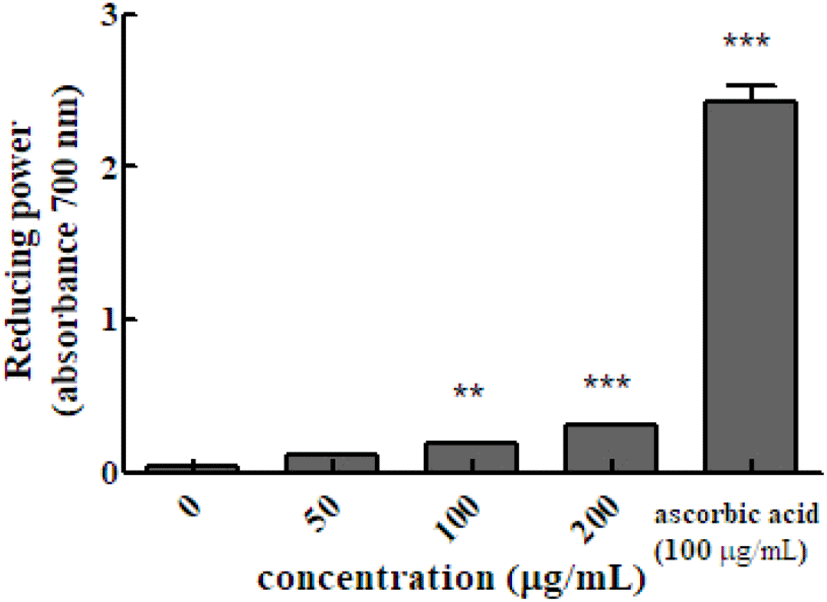
Reducing power is determined by measuring the intensity of blue Perl’s Prussian blue at 700 nm, which is due to the reduction of the Fe3+/ferricyanide complex (potassium ferricyanide (III)) by antioxidants into the ferrous form. The absorbance value indicates the reducing power of the sample. Higher the absorbance value, indicate increase reducing power. The absorbance value the of hot water extract of Em leaves at concentrations of 50, 100, and 200 μg/mL was 0.118±0.002, 0.185±0.006, and 0.312±0.002, respectively, which were higher than that of the control group (100 μg/mL ascorbic acid, 2.196±0.027).
Phenolic compounds exhibit a high antioxidant activity as secondary metabolites and are widely distributed in nature. The hydroxyl group of an aromatic compound binds to phenolic compounds. Thus, when exposed to active oxygen, phenolic compounds exhibit antioxidant and anti-cancer activities. They also protect DNA, cellular constituents, and enzymes from damage. The total phenolic content of the hot water extract of Em leaves was 31.687±4.904 TAE/g, and the leaves of Em are considered to have higher antioxidant activity than the fruits (Fig. 2). The polyphenol content in the leaves has been reported to be 805.6 mg/100 g (Yoon et al., 2007).
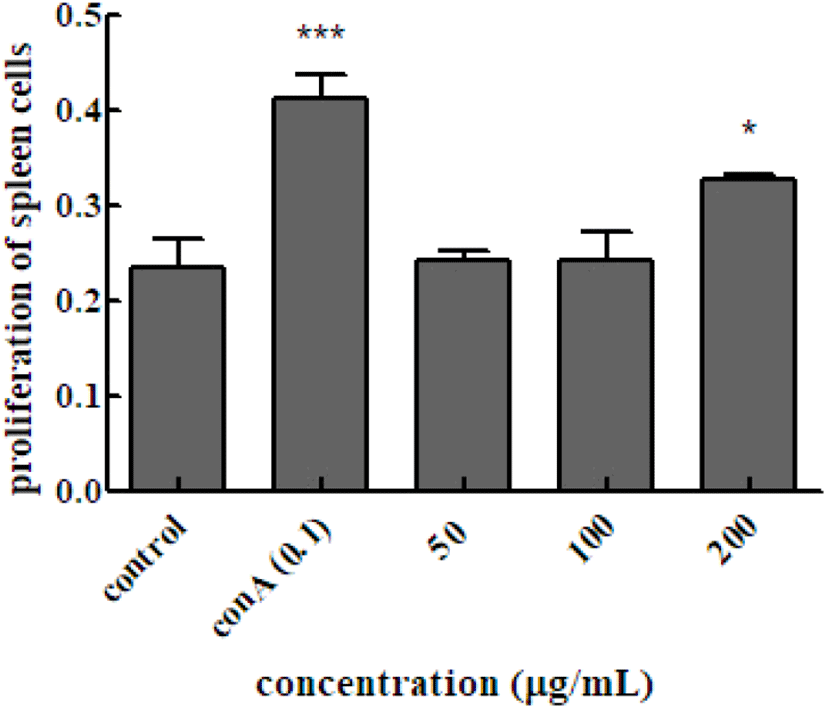
To investigate the effect of Em leaf extracts on ConAinduced responses in splenocytes, splenocytes were isolated from mice and cultured by adding only the Em leaf extract to the culture medium. As shown in Fig. 3, the absorbance value of the group cultured with the ConA mitogen was higher than that of the group cultured without the mitogen. It was also confirmed that splenocyte proliferation was increased. The absorbance of the cell suspension with the Em leaf extract at different concentrations (50, 100, and 200 μg/mL), measured after 48 h of treatment increased (0.241± 0.011, 0.242±0.031, and 0.328±0.004, respectively) compared to the value of 0.233±0.030 in the extract-free splenocyte control. The increase in absorbance in the 200 μg/mL treatment group was 1.5-fold higher than that in the control group.
We next evaluated the effect of the hot water leaf extract of Em on the ability of spleen cells to produce the IL-2, IL-4, and IL-10 cytokines following induction by ConA. Spleen cells of Balb/c mice were treated with different concentrations of the extract and cultured for 48 h. The levels of IL-2, IL-4, and IL-10 in the culture were measured.
In the immune response, IL-2 is important for signal transduction between cells in the initial inflammatory responses (Barnes and Liew, 1995). IL-2 is widely used as an indicator of enhanced immune response (Grabstein et al., 1994). To confirm the immune-enhancing effect, we measured the level of IL-2, a Th1 cytokine secreted by mouse spleen Th cells. IL-2 is secreted by activated T cells and is primarily a cytokine that primarily acts as a T-cell growth factor by promoting T-cell division. Increased level of T lymphocytes can be used as an index of the effect of IL-2. Spleen cells treated with ConA, which activates only T cells, and those treated with only the extract were cultured for 48 h, and then IL-2 levels were measured. The IL-2 level was 2.298±0.321 in the control group and 1.451± 0.149, 1.583±0.133, and 1.855±0.171 pg/ml in the 50, 100, and 200 μg/mL treatment groups, respectively (Fig. 4A).
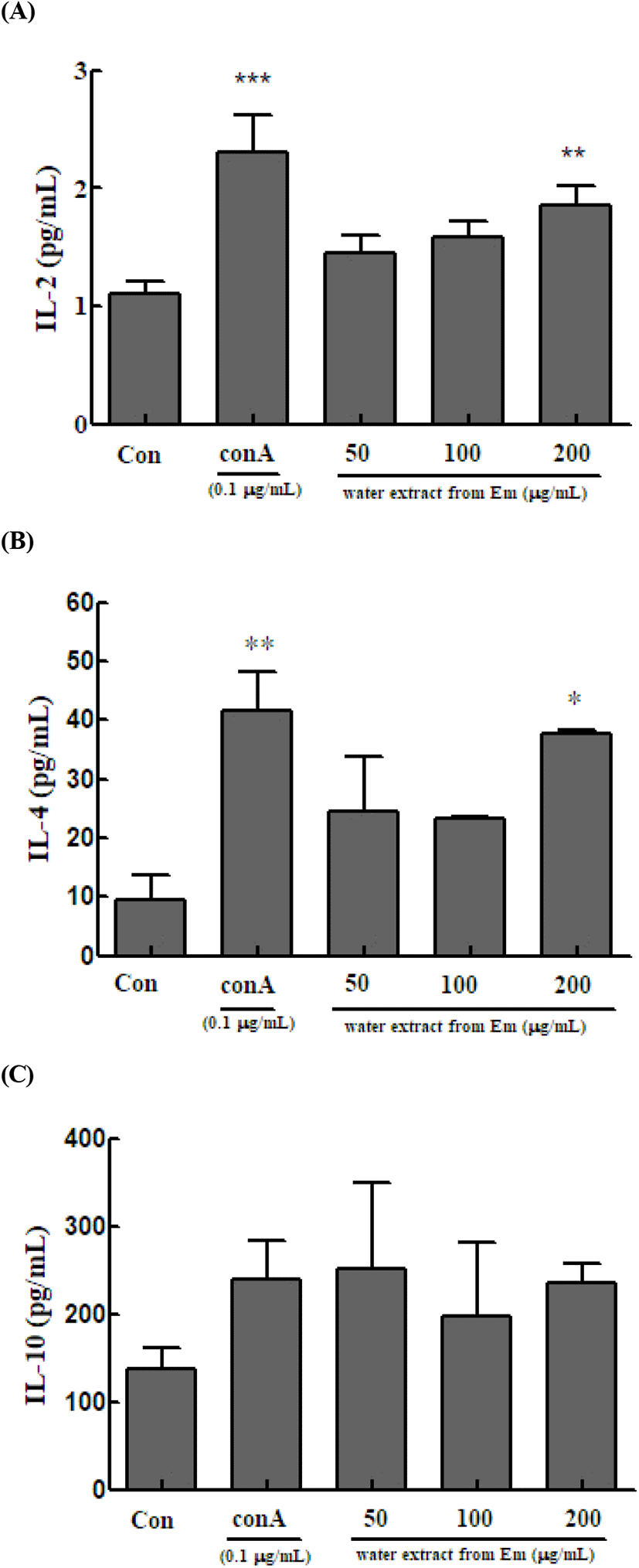
Th2 cells play a major role in humoral immunity by producing antibodies and suppressing the action of Th1 cells. In the immune system, Th2 cells maintain homeostasis due to the Th1/Th2 cell balance (Seo et al., 2013). We measured IL-4 and IL-10 in Th2 cells. IL-4 is secreted by T, B, and NK cells, monocytes, macrophages, neutrophils, eosinophils, vascular endothelial cells, and fibroblasts. It targets and induces the differentiation and proliferation of Th2 cells and simultaneously increases the number of CD4 + T cells (Seo et al., 2013). Spleen cells treated with ConA and those treated with only the extract were cultured for 48 h, and then IL-4 levels were measured. The IL-4 level was 41.574±6.678 in the control group and 24.556±9.219, 23.407±0.262, and 37.630±0.576 pg/mL in the 50, 100, and 200 μg/mL treatment groups, respectively (Fig. 4B).
IL-10 targets monocytes, macrophages, T cells, B cells, NK cells, and mast cells. It is an anti-inflammatory cytokine that acts directly on T cells and macrophages to suppress cell activation (Yi et al., 2013) and is an inhibitory cytokine that suppresses the immune system (Sohn et al., 2012). Spleen cells treated with ConA and those treated with only the extract were cultured for 48 h, and then IL-10 levels were measured. The IL-10 level in the control cells was 239.111±43.299 μg/mL, whereas that in the treatment groups (50, 100, and 200 μg/mL) was 252.250±96.284, 196.500±84.297, and 236.167±22.156 μg/mL, respectively (Fig. 4C).
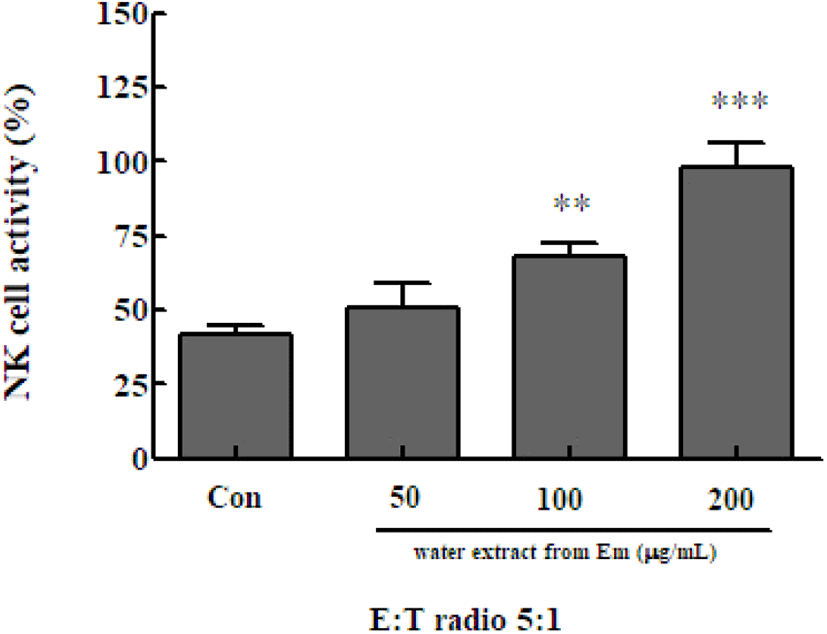
NK cells secrete water soluble factors, including cytokines, that can directly kill cancer cells and cells infected by intracellular microorganisms. NK cells can also activate other predatory cells, such as monocytes and dendritic cells. Immune cells play a major role in innate immunity (Song et al., 2008). To determine the NK cell activity following treatment with the Em leaf extract, NK cells were isolated from spleen cells and co-cultured with Yac-1 mouse lymphoma cells. Cell-killing ability was evaluated using the Cytotox 96 non-radioactive cytotoxicity assay kit. NK cell activity was significantly increased in the presence of 100 μg/mL (67.874±4.877%) and 200 μg/mL (97.975±8.598%) of the Em extract, compared with activity in the extract-free control group (41.985±2.604%)(Fig. 5). The activity of NK cells of the treatment group was significantly higher than that of the control group at all extract concentrations.